

中国科学院物理研究所
北京凝聚态物理国家研究中心
N04组供稿
第28期
2010年12月16日
固体表面上功能分子体系组装构造的动态过程及其调控研究取得新进展
功能分子在固体表面上的可控组装及其物性研究,一直是纳米器件量子调控的重要研究内容。目前,分子在表面自组装方面的研究主要是通过设计分子结构,调制分子-基底以及分子-分子间相互作用,从而得到所需的功能特性。其中,分子在表面动态过程中的表征一直是富有挑战性的问题,特别是对于结构较为复杂的大分子复杂体系,目前尚无对于整个动态体系进行全面描述的方法。中科院物理研究所/北京凝聚态物理国家实验室(筹)高鸿钧研究员领导的研究团队近年来一直致力于功能分子在固态表面上的生长、动态行为及其物理性质方面的研究(Phys. Rev. Lett. 97, 156105 (2006);96, 226101 (2006);96, 156102 (2006);99, 106402 (2007);100, 186104 (2008)),取得了一系列创新性的成果,例如在构筑实用化单分子转子及其阵列方面,首次实现了具有固定偏心轴的单分子转子的大面积组装(Phys. Rev. Lett. 101,197209(2008))等等。
最近该研究组的刘奇等提出了一种研究复杂分子表面动态系统的新方法:利用时间分辨的隧穿电流谱(I-t)来获取表面动态过程中复杂分子的构型和能态分布。针对表面分子动态系统当前的研究瓶颈(包括运动过程中分子的多种构型的识别和对动态系统进行时间分辨和空间分辨的详细描述), 他们通过液氮温度下两个典型的表面动态体系: Au(111) 表面的 (t-Bu)4-ZnPc分子马达体系和Au(111) 表面的FePc分子流体体系,利用时间分辨的隧穿电流谱结合第一性原理计算,成功确定了体系中分子的多种不同构型和能态分布(参见图1),并且首次通过时间分辨电流谱及其提出的分析方法,获得了FePc在Au(111) 表面上不同位置处吸附能差的分布曲面(参见图2)。上述结果对于表面动态系统的研究提出了一种新的研究思路,成功地解决了目前在分子特别是复杂分子动态体系研究中存在的困难问题,该方法也可应用于相关领域的研究。相关研究结果发表在Phys. Rev. Lett. 104, 166101 (2010)上。
与此同时该研究组的江楠等深入研究了有机分子-金属体系中分子扩散动态行为的电场调控及其内在原理机制。他们使用低温STM在液氮温度(~77K)下对FePc分子在Au (111) 表面的扩散现象进行了详细表征,并且在可控外加电场的作用下,对于分子在表面的运动和静止进行了调控,实现了超分子结构可控可逆的分解与再组装。他们同时结合DFT理论计算,详细分析了外加电场对分子和基底间相互作用的控制以及对分子在表面扩散势垒的影响。这项成果的意义在于将人为的可控设计和自发的分子自组装特性结合在一起,为设计和构筑纳米尺度下的分子电子器件提供了新的途径(参见图3)。美国Vanderbilt大学S. Pantelides教授研究组进行了该工作的部分理论与计算方面的合作。相关研究结果发表在Nano Letters. 10, 1184 (2010)上。
在理解分子与基底的精细作用机制方面,德国一研究团队试图将色散力(dispersion force)的重要性推广到有机分子-金属表面,他们认为吡啶等杂环有机分子和Cu表面的主要相互作用力是色散力(Atodiresei et al., Phys. Rev. Lett. 102, 136809 (2009))。通过全面的理论计算与模拟,加拿大的季威博士(先前物理所N04组博士生)、西班牙L.A. Zotti教授、英国W.A. Hofer教授与高鸿钧研究员等发现Atodiresei等人是由于采用了一个不稳定的构型,从而得出了上述不准确的结论。在分别计算了平行和垂直表面的两种能量最低构型的电子结构后,季威等发现这些分子与表面的主要相互作用机制是典型的化学相互作用,即共价键,而非色散力。这一结果为正确描述分子-金属表面的精细相互作用机制给出了重要结论。相关结果发表在Phys. Rev. Lett. 104, 099703 (2010)上。
上述工作是在中国科学院、科技部、国家自然科学基金委的支持下完成。
附件:
1. Q. Liu, Y. Y. Zhang, N. Jiang, H. G. Zhang, L. Gao, S. X. Du, and H. -J. Gao, "Identifying Multiple Configurations of Complex Molecules in Dynamical Processes: Time Resolved Tunneling Spectroscopy and Density Functional Theory Calculation"
Phys. Rev. Lett. 104, 166101 (2010).
2. N. Jiang, Y. Y. Zhang, Q. Liu, Z. H. Cheng, Z. T. Deng, S. X. Du, H. -J. Gao, M. J. Beck and S. T. Pantelides, "Diffusivity Control in Molecule-on-Metal Systems Using Electric Fields"
Nano Letters 10. 1184-1188 (2010).
3. W. Ji, L. A. Zotti, H.-J. Gao, and W. A. Hofer, Comment on “Chemical versus van der Waals Interaction: The Role of the Heteroatom in the Flat Absorption of Aromatic Molecules C6H6, C5NH5, and C4N2H4 on the Cu(110) Surface”
Phys. Rev. Lett. 104, 099703 (2010).
最近该研究组的刘奇等提出了一种研究复杂分子表面动态系统的新方法:利用时间分辨的隧穿电流谱(I-t)来获取表面动态过程中复杂分子的构型和能态分布。针对表面分子动态系统当前的研究瓶颈(包括运动过程中分子的多种构型的识别和对动态系统进行时间分辨和空间分辨的详细描述), 他们通过液氮温度下两个典型的表面动态体系: Au(111) 表面的 (t-Bu)4-ZnPc分子马达体系和Au(111) 表面的FePc分子流体体系,利用时间分辨的隧穿电流谱结合第一性原理计算,成功确定了体系中分子的多种不同构型和能态分布(参见图1),并且首次通过时间分辨电流谱及其提出的分析方法,获得了FePc在Au(111) 表面上不同位置处吸附能差的分布曲面(参见图2)。上述结果对于表面动态系统的研究提出了一种新的研究思路,成功地解决了目前在分子特别是复杂分子动态体系研究中存在的困难问题,该方法也可应用于相关领域的研究。相关研究结果发表在Phys. Rev. Lett. 104, 166101 (2010)上。
与此同时该研究组的江楠等深入研究了有机分子-金属体系中分子扩散动态行为的电场调控及其内在原理机制。他们使用低温STM在液氮温度(~77K)下对FePc分子在Au (111) 表面的扩散现象进行了详细表征,并且在可控外加电场的作用下,对于分子在表面的运动和静止进行了调控,实现了超分子结构可控可逆的分解与再组装。他们同时结合DFT理论计算,详细分析了外加电场对分子和基底间相互作用的控制以及对分子在表面扩散势垒的影响。这项成果的意义在于将人为的可控设计和自发的分子自组装特性结合在一起,为设计和构筑纳米尺度下的分子电子器件提供了新的途径(参见图3)。美国Vanderbilt大学S. Pantelides教授研究组进行了该工作的部分理论与计算方面的合作。相关研究结果发表在Nano Letters. 10, 1184 (2010)上。
在理解分子与基底的精细作用机制方面,德国一研究团队试图将色散力(dispersion force)的重要性推广到有机分子-金属表面,他们认为吡啶等杂环有机分子和Cu表面的主要相互作用力是色散力(Atodiresei et al., Phys. Rev. Lett. 102, 136809 (2009))。通过全面的理论计算与模拟,加拿大的季威博士(先前物理所N04组博士生)、西班牙L.A. Zotti教授、英国W.A. Hofer教授与高鸿钧研究员等发现Atodiresei等人是由于采用了一个不稳定的构型,从而得出了上述不准确的结论。在分别计算了平行和垂直表面的两种能量最低构型的电子结构后,季威等发现这些分子与表面的主要相互作用机制是典型的化学相互作用,即共价键,而非色散力。这一结果为正确描述分子-金属表面的精细相互作用机制给出了重要结论。相关结果发表在Phys. Rev. Lett. 104, 099703 (2010)上。
上述工作是在中国科学院、科技部、国家自然科学基金委的支持下完成。
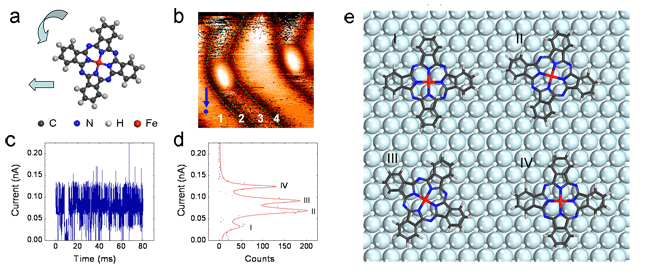 |
| 图1:Au(111)面上的酞菁铁流体体系的时间分辨隧穿电流谱(I-t)谱及统计结果分析得到的能态分布。 (a) 酞菁铁分子结构。(b)为液氮下的酞菁铁的STM图像,蓝色圆点表明了进行I-t谱测量的位置,在Au(111)面上的FCC区域。(c)和(d)是所获得的I-t谱及其统计曲线,有两个能量非常接近的构型II和III,它们的占据几率接近70%, II和III构型的分子取向是一致的,这表明在FCC区域,酞菁铁分子的扩散主要表现为平移。(e)标示出了第一性原理计算出的与(d)对应的四种构型。 |
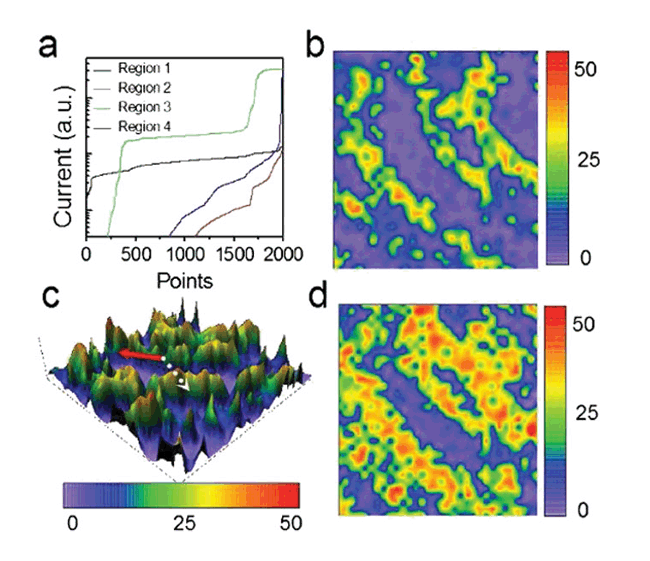 |
| 图2:基于I-t谱统计结果分析与第一性原理计算的测量表征方法,可直接得到表面吸附能的空间分布和能量分布。(a)是图1b中标示出的四个不同测量点所对应的排序I-t谱(对原始I-t谱进行从小到大排序,然后对于纵轴的电流值采用对数坐标),它们具有不同的形状。( b)是E1的空间分布图案,E1为最稳定构型和次稳定构型间的能量差。(c)是(b)的三维表示,实线箭头和虚线箭头标明了不同扩散方向上的能量区别,在中间的金条纹突起上,能量差别变得比较大,说明分子的构型转变难以在这些区域进行,因此扩散受到限制。(d)是E2的空间分布图案,E2为最稳定构型和第三稳定构型间的能量差。此图案表明在FCC中的某些区域,第三稳定构型同第一稳定构型间的能量区别不大,在这些区域,分子可以进行更多种的构型改变,包括平移、平面旋转等等多种自由度的运动。图中的彩色条单位是meV。 |
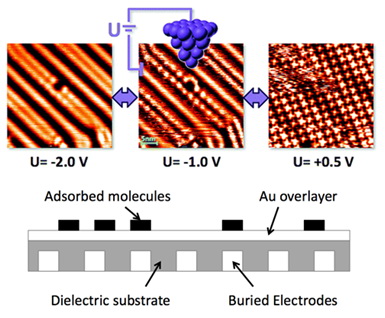 |
| 图3:酞菁铁(FePc)分子在Au(111)表面的运动和静止状态的转变。利用外加电场调制了分子在基底表面的扩散能力,可控的实现分子在表面的运动和静止,使得表面分子自组装结构产生巨大又可逆的变化。如果把FePc-Au体系放在预埋电极的绝缘基底上,可搭建一个受电场控制的可擦写分子器件。 |
1. Q. Liu, Y. Y. Zhang, N. Jiang, H. G. Zhang, L. Gao, S. X. Du, and H. -J. Gao, "Identifying Multiple Configurations of Complex Molecules in Dynamical Processes: Time Resolved Tunneling Spectroscopy and Density Functional Theory Calculation"
Phys. Rev. Lett. 104, 166101 (2010).
2. N. Jiang, Y. Y. Zhang, Q. Liu, Z. H. Cheng, Z. T. Deng, S. X. Du, H. -J. Gao, M. J. Beck and S. T. Pantelides, "Diffusivity Control in Molecule-on-Metal Systems Using Electric Fields"
Nano Letters 10. 1184-1188 (2010).
3. W. Ji, L. A. Zotti, H.-J. Gao, and W. A. Hofer, Comment on “Chemical versus van der Waals Interaction: The Role of the Heteroatom in the Flat Absorption of Aromatic Molecules C6H6, C5NH5, and C4N2H4 on the Cu(110) Surface”
Phys. Rev. Lett. 104, 099703 (2010).