

中国科学院物理研究所
北京凝聚态物理国家研究中心
N04组供稿
第93期
2018年12月29日
金属表面有机分子对称性破缺诱导的选择性功能化
近年来,将第一性原理计算与扫描隧道显微镜(STM)和原子力显微镜(AFM)实验相结合已成为在原子、分子层次研究表面物理和化学过程的强有力手段,在实现小分子甚至单原子级别的操纵和表面化学反应的基础上,可以进一步研究原子尺度下的新奇物理化学性质。
表面合成是近年来备受关注的一种合成方法。利用金属单晶表面的催化和限域效应,合成出了大量传统湿法化学方法所无法合成的低维功能性纳米材料。中国科学院物理研究所/北京凝聚态物理国家研究中心杜世萱研究员等在这一领域取得了一系列居国际前沿的研究成果,2014年,他们将第一性原理计算与STM实验相结合,首次证实在Au表面发生的炔类分子的环化三聚加成反应为一个[2 + 2 + 2]的两步反应,同时发现表面限域效应有效提高了反应的选择性,相关工作发表在J. Am. Chem. Soc. 136, 5567 (2014)上。今年,他们与德国明斯特大学H. Fuchs教授课题组合作,发现了不同金属基底对表面乌尔曼反应的调制作用,相关工作发表在Chem. Commun. 54, 9305 (2018)上;此外,他们与劳伦斯伯克利国家实验室合作研究了表面金属有机纳米颗粒和纳米晶体自组装形成二维超结构的机理,相关工作发表在Angew. Chem. Int. Ed. 57, 13172 (2018)上。这些精细结构的合成表明,衬底表面以及功能分子的自身结构等在不同官能团的活化和成断键中具有精准的选择性。
有机分子的可控选择性功能化对在原子尺度实现低维材料精准构造具有重要意义。然而,实现有机分子中相似基团的选择性活化,特别是对称分子中等价基团的选择性活化,是传统化学合成中的一个历史性难题。
最近,杜世萱研究员(共同通讯作者)与苏州大学迟力峰教授和德国吉森大学André Schirmeisen教授合作,通过4,4″二氨基对三联苯(DATP)分子在Cu(111)表面上吸附导致的对称性破缺,实现了DATP中两个相同氨基的选择性活化。镜面对称的DATP分子在Cu(111)表面上吸附时,分子长轴沿着[11-2]及其等价晶向方向,在AFM图像中分子一端呈现模糊的特征,分子的镜面对称被打破(图1)。化学键分辨的AFM表征结合第一性原理计算揭示了DATP分子与Cu(111)的晶格失配使得分子内两个等价的氨基官能团吸附在铜表面的不同位置,一端吸附在铜原子的顶位,另一端吸附在表层Cu原子的空心位(图3)。其中吸附在顶位的氨基相比另一端的氨基与铜表面的距离更近,与表面的相互作用更强,因此活化程度更强,使其在针尖的影响下呈现非稳态特征,并且优先与2-三亚苯甲醛(TPCA)分子以氢键结合(图5)。当将DATP分子吸附在Au(111)表面上时,由于分子长度与基底晶格匹配,分子仍然保持镜面对称,两端的氨基吸附位置相同,因此两端氨基的活化程度相同(图4)。DATP分子与Cu(111)表面的晶格失配引起的分子不对称吸附和特定官能团的活性增强,为对称分子中等价基团的选择性功能化提供了一种广泛使用的途径,为表面上的不对称化学反应提供了一条新的思路。
相关工作发表在Nature Communication 9, 3277 (2018)上, 该工作得到了国家自然科学基金中德合作计划、科技部和中国科学院的支持。
文章链接:https://www.nature.com/articles/s41467-018-05719-y
表面合成是近年来备受关注的一种合成方法。利用金属单晶表面的催化和限域效应,合成出了大量传统湿法化学方法所无法合成的低维功能性纳米材料。中国科学院物理研究所/北京凝聚态物理国家研究中心杜世萱研究员等在这一领域取得了一系列居国际前沿的研究成果,2014年,他们将第一性原理计算与STM实验相结合,首次证实在Au表面发生的炔类分子的环化三聚加成反应为一个[2 + 2 + 2]的两步反应,同时发现表面限域效应有效提高了反应的选择性,相关工作发表在J. Am. Chem. Soc. 136, 5567 (2014)上。今年,他们与德国明斯特大学H. Fuchs教授课题组合作,发现了不同金属基底对表面乌尔曼反应的调制作用,相关工作发表在Chem. Commun. 54, 9305 (2018)上;此外,他们与劳伦斯伯克利国家实验室合作研究了表面金属有机纳米颗粒和纳米晶体自组装形成二维超结构的机理,相关工作发表在Angew. Chem. Int. Ed. 57, 13172 (2018)上。这些精细结构的合成表明,衬底表面以及功能分子的自身结构等在不同官能团的活化和成断键中具有精准的选择性。
有机分子的可控选择性功能化对在原子尺度实现低维材料精准构造具有重要意义。然而,实现有机分子中相似基团的选择性活化,特别是对称分子中等价基团的选择性活化,是传统化学合成中的一个历史性难题。
最近,杜世萱研究员(共同通讯作者)与苏州大学迟力峰教授和德国吉森大学André Schirmeisen教授合作,通过4,4″二氨基对三联苯(DATP)分子在Cu(111)表面上吸附导致的对称性破缺,实现了DATP中两个相同氨基的选择性活化。镜面对称的DATP分子在Cu(111)表面上吸附时,分子长轴沿着[11-2]及其等价晶向方向,在AFM图像中分子一端呈现模糊的特征,分子的镜面对称被打破(图1)。化学键分辨的AFM表征结合第一性原理计算揭示了DATP分子与Cu(111)的晶格失配使得分子内两个等价的氨基官能团吸附在铜表面的不同位置,一端吸附在铜原子的顶位,另一端吸附在表层Cu原子的空心位(图3)。其中吸附在顶位的氨基相比另一端的氨基与铜表面的距离更近,与表面的相互作用更强,因此活化程度更强,使其在针尖的影响下呈现非稳态特征,并且优先与2-三亚苯甲醛(TPCA)分子以氢键结合(图5)。当将DATP分子吸附在Au(111)表面上时,由于分子长度与基底晶格匹配,分子仍然保持镜面对称,两端的氨基吸附位置相同,因此两端氨基的活化程度相同(图4)。DATP分子与Cu(111)表面的晶格失配引起的分子不对称吸附和特定官能团的活性增强,为对称分子中等价基团的选择性功能化提供了一种广泛使用的途径,为表面上的不对称化学反应提供了一条新的思路。
相关工作发表在Nature Communication 9, 3277 (2018)上, 该工作得到了国家自然科学基金中德合作计划、科技部和中国科学院的支持。
文章链接:https://www.nature.com/articles/s41467-018-05719-y
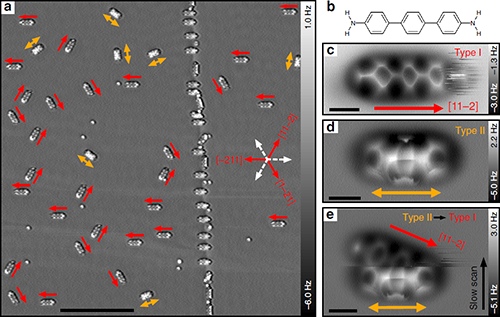 |
| 图1. DATP分子在Cu(111)表面的两种吸附构型。 |
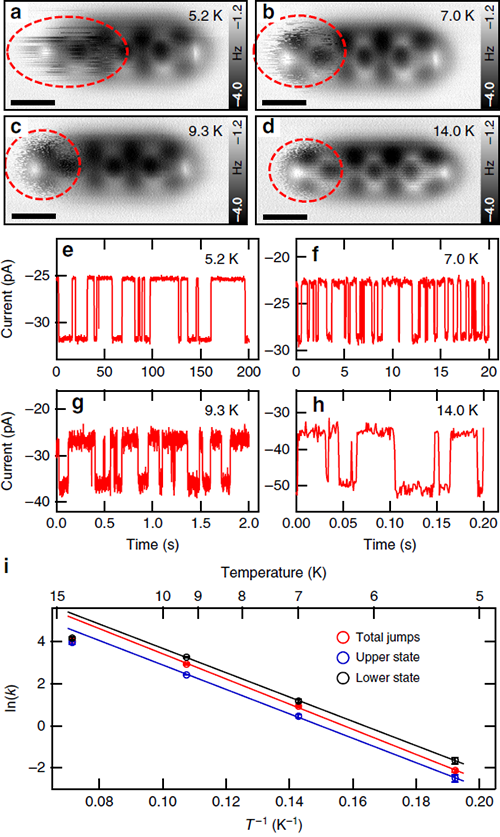 |
| 图2. 观测到的跳跃机制的动力学分析和能量势垒。 |
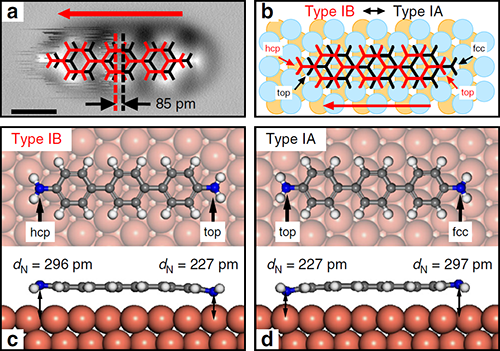 |
| 图3. DATP分子在Cu(111)表面的第一种不对称的吸附构型。 |
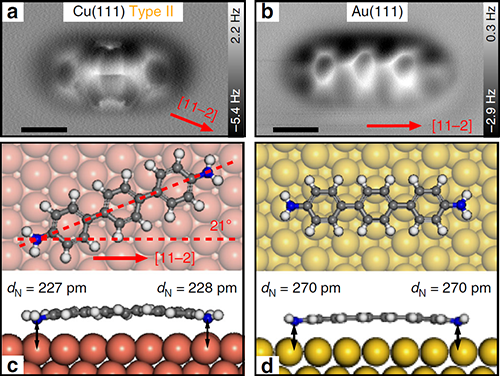 |
| 图4. DATP分子在Cu(111)和Au(111)表面上的第二种对称的吸附构型。 |
 |
| 图5. DATP和TPCA分子的自组装。 |